Eventos
Avances terapéuticos en Venezuela en la atención de las enfermedades raras

Esta iniciativa dirigida a un reducido grupo de periodistas científicos, estuvo precedida por la reconocida experticia de la Dra. Antonieta Mahfoud, Médico con Postgrado en Pediatría y Neurología Infantil. Experto en errores innatos del metabolismo, perteneciente al Programa de Estimulo al Investigador desde 2004. Directora del Área de Ciencia y Biotecnología para la Salud del Instituto de Estudios Avanzados IDEA, donde se desempeña como Neuropediatra desde 1996. Directora de la Sección de Neuropediatría de la Sociedad Venezolana de Neurología 2010-2012. Miembro titular de la Sociedad Latinoamericana de Errores Innatos del Metabolismo y Pesquisa Neonatal desde 1999. Posee más de 20 publicaciones en revistas Nacionales e Internacionales. Me permito estimados lectores, ofrecerles un resumen de su presentación, en el aspecto clínico de éstas patologías.
Por: Julio César Alcubilla
Mail: [email protected] y [email protected]
twitter@editorglobal
"A estas enfermedades muchas veces las identificamos como un enigma médico, debido a que no las logramos dilucidar y cuando las alcanzamos, ya son determinantes. Las enfermedades raras son potencialmente mortales o debilitantes, al ser raras son de muy baja prevalencia y además muestran dificultades para llegar a su diagnóstico, por ser de un alto nivel de complejidad. Algunas definiciones coinciden que son enfermedades que tienen 5 o menos casos por cada 10.000 habitantes, en Venezuela pudiésemos tener 576.000, en tal sentido, las enfermedades raras son minoritarias, pero sus pacientes numerosos".
"Tres de cada cuatro enfermedades raras se presentan en la infancia y determinan discapacidades graves, causando el 35% de las muertes en menores de 1 año. Siendo el 80% por errores de origen genético, afectan al sistema nervioso en un 44.93% de los pacientes, metabólicas hereditarias en un 41.01%, al aparato locomotor en un 30,72% y al tejido conectivo en un 24,35%. Por ello se hace necesario tomarlas en cuenta, como problema de salud pública".
"Estas enfermedades analizando su entorno terapéutico y la situación de medicamentos en nuestro país y a escala mundial, existe una escasa disponibilidad de los mismos por su alto costo, muchos de los pacientes solo tienen tratamiento sintomático además de requerir atención y seguimiento multidisciplinario. Sin embargo, la investigación y la ciencia de la biotecnología llevada a cabo en nuestro país, por el Instituto de Estudios Avanzados IDEA, ha permitido el desarrollo de terapias que mejoran la esperanza y calidad de vida de los pacientes afectados, tales como: La Terapia de Reemplazo Enzimático, el uso de chaperonas, la terapia génica y la Nanotecnología. Normalmente un médico si no hace un estudio adecuado, no logra un diagnóstico efectivo, se requieren estudios especializados para llegar a óptimos resultados".

"Por ello el entrenamiento en diagnóstico es un área vital y estratégica, debido a que el diagnóstico temprano va a condicionar al tratamiento, el cual tiene un gran impacto para prolongar la vida del paciente. En cuanto a la situación psicosocial de estos pacientes, al revisar toda la literatura existente, estas enfermedades además de ser enigmáticas, son las grandes simuladoras, se confunden con muchas otras. Para su diagnóstico los especialistas deben ser muy acuciosos, por ello se requiere educación médica continua, hasta que se llegue algún día al médico que va a asistir al paciente en atención primaria. Se ha de entender, que el paciente al llegar al especialista ya ha recorrido un gran camino. ¿Cómo se puede ver la situación socio sanitaria de éstos pacientes?, definitivamente desde distintas dimensiones hasta llegar a un todo, aportando una solución integral a su realidad. Como lo refleja la Organización Mundial de la Salud (OMS), ver a estos pacientes como un valor vivo que ha de ser atendido. La lectura ecuménica de los pacientes es necesaria, analizarlos en todas sus dimensiones eliminará las barreras que impiden la participación plena. Estos pacientes tienen enfermedades que los llevan a discapacitarlos, para integrarse en todos los ámbitos del medio escolar, su familia y la sociedad".
"En tal sentido los errores innatos del metabolismo, en cuanto a sus generalidades, al aislar y revisar la frecuencia de cada una de estas enfermedades, se logran constatar que sus manifestaciones pueden afectar cualquier órgano o sistema. Se definen con orígenes genéticos, lo cual normalmente representa una mutación de la proteína, que no va a funcionar o trabajar correctamente. Ocasionando un déficit energético, el mismo se puede manifestar como: encefalopatía severa sin intervalo, libre de síntomas, el paciente presenta Hipotonía y Convulsiones. O pudiese presentarse con Encefalopatía con afectación multisistémica progresiva, que incluye dismorfias y cardiomiopatías. Trayendo como consecuencia enfermedades del peroxisoma, defectos b-oxidación de ácidos grasos, defectos de neurotransmisores o Enfermedades Mitocondriales".
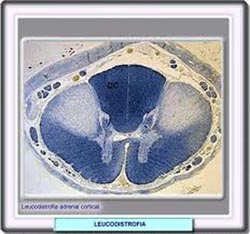 "Al analizar las enfermedades del peroxisoma, sus manifestaciones pueden ser permanentes y progresivas y a su vez pueden presentarse síntomas variados y neurológicos. Entre los síntomas variados destacan: cataratas, opacidad corneal y alteraciones del aparato respiratorio. En lo referente a los síntomas neurológicos: Hipotonía, Macrocefalia o Hidrocefalia. Por su parte, la disfunción de las moléculas complejas, define un trastorno en la síntesis o catabolismo de moléculas complejas, que trae como consecuencia a los pacientes manifestaciones permanentes y progresivas. Presentándose por igual, síntomas variados y neurológicos: entre los síntomas variados destacan: cataratas u opacidad corneal, alteraciones del aparato respiratorio, afectación cardíaca, Hepatoesplenomegalia, Hernias, Facies o facciones toscas, alteraciones. En lo referente a los trastornos o síntomas neurológicos: Hipotonía, Macrocefalia/Hidrocefalia, retardo psicomotor, Epilepsia, trastornos conductuales, deterioro mental progresivo y Compresión Medular".
"Al analizar las enfermedades del peroxisoma, sus manifestaciones pueden ser permanentes y progresivas y a su vez pueden presentarse síntomas variados y neurológicos. Entre los síntomas variados destacan: cataratas, opacidad corneal y alteraciones del aparato respiratorio. En lo referente a los síntomas neurológicos: Hipotonía, Macrocefalia o Hidrocefalia. Por su parte, la disfunción de las moléculas complejas, define un trastorno en la síntesis o catabolismo de moléculas complejas, que trae como consecuencia a los pacientes manifestaciones permanentes y progresivas. Presentándose por igual, síntomas variados y neurológicos: entre los síntomas variados destacan: cataratas u opacidad corneal, alteraciones del aparato respiratorio, afectación cardíaca, Hepatoesplenomegalia, Hernias, Facies o facciones toscas, alteraciones. En lo referente a los trastornos o síntomas neurológicos: Hipotonía, Macrocefalia/Hidrocefalia, retardo psicomotor, Epilepsia, trastornos conductuales, deterioro mental progresivo y Compresión Medular"."Los análisis o estudios necesarios para determinar éstas enfermedades, los complementan: la historia clínica, análisis bioquímicos, estudios enzimáticos y estudios moleculares. En su fase clínica, cuando se sospecha una Enfermedad de Errores Innatos del Metabolismo (EIM), se observa rápido deterioro de neonato o lactante después de un período libre de síntomas. Vómitos, Estenosis Pilórica, Vómitos recurrentes con letargia, Coma, Ataxia aguda, Estridor laríngeo- déficit de biotinidasa. Entre sus causas pudiesen hallarse antecedentes familiares como la consanguinidad parental, el origen étnico o geográfico, abortos a repetición, muertes neonatales o infantiles tempranas sin causas o inexplicadas, o un caso anterior en la familia".
"Dentro de las enfermedades raras, existe un grupo llamadas Enfermedades de Depósito Lisosomal, entre las cuáles se han identificado 60 hasta la fecha y solo 6 de ellas tienen tratamiento: Pompe, MPSI, MPSII, MPSIV, Fabry y Gaucher. Para estas enfermedades se encuentran tratamientos denominados Terapia de Reemplazo Enzimático.
Manifestaciones Clínicas
Enfermedad de POMPE: EDL caracterizada por debilidad muscular que se manifiesta principalmente en los músculos esqueléticos, respiratorios del corazón, se produce por la deficiencia de la enzima Lisosomal Alfa Glucosidasa Ácida.
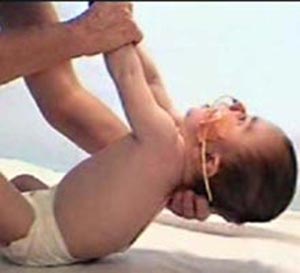
| Enfermedad de POMPE |
Mucopolisacaridosis (MPS): EDL que se caracteriza por acúmulo de carbohidratos complejos llamados Glucosaminoglanos a nivel Lisosomal, los cuáles se acumulan en cantidades anormales en múltiples órganos y tejidos. Existen descritas 7 subtipos de MPS, dentro de los cuáles el Síndrome de Hunter (MPS I), pudiese pasar desapercibida o mal diagnosticada por años, debido a su presentación sutil y poco específica de los síntomas musculoesqueléticos. Los pacientes presentan síntomas similares a la artritis; es también de herencia autosómica recesiva y existe una deficiencia de la enzima Alfa- L- iduronidasa.
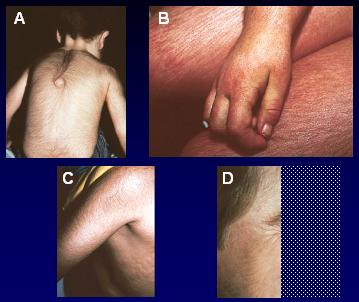
| Mucopolisacaridosis (MPS) |
Enfermedad de Gaucher: Es una enfermedad hereditaria causada por la deficiencia de la enzima Lisosomal Glucocerebrosidasa. Posee un patrón de herencia autosómica recesiva. Propicia la alteración del sistema inmunológico.
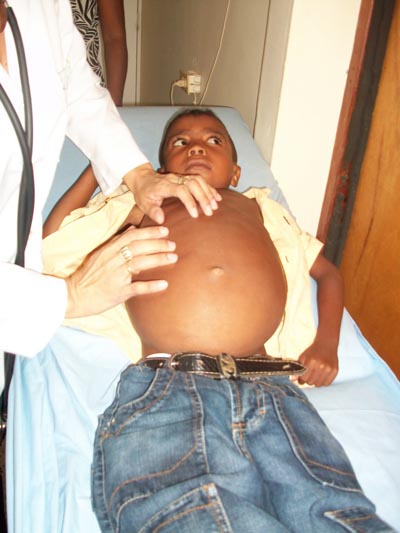
| Enfermedad de Gaucher |
Enfermedad FABRY: EDL causada por la deficiencia de la enzima Lisosomal Alfagalactosidasa A. Cursa patrón hereditario ligado al Cromosoma X: mujeres portadoras asintomáticas, hombres manifiestan la enfermedad. Se produce afectación del endotelio vascular y puede producir graves complicaciones renales, cardíacas y cerobrovasculares.
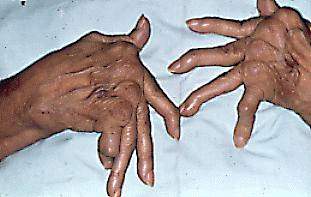
| Enfermedad de FABRY |
Dra. Antonieta Mahfoud
Fuente: Julio César Alcubilla – TecnologiaHechaPalabra
