Distrofia muscular progresiva: características, tratamiento y prevención
• La incidencia promedio es 1 en 3.500 varones nacidos vivos. Es el tipo más frecuente y severo de Distrofia Muscular en niños.
• Representa uno de los mayores problemas o retos para Pediatras y Neurólogos Infantiles en el Asesoramiento Médico Integral, así como para los Genetistas Clínicos.
• En años recientes han habido avances importantes en la etiopatogenia, diagnóstico, tratamiento y en la atención especializada de los pacientes.
Características clínicas
– Se presenta en varones, y hay variabilidad en la expresión clínica
– Comienza generalmente de 3 a 5 años, después que el niño empezó a caminar independientemente.
– En ocasiones, inicialmente se observan pies planos, pies evertidos, o los pacientes caminan en la punta de los pies.
– Creciente dificultad para subir escaleras, cansancio al caminar o correr, caídas frecuentes y contracturas musculares.
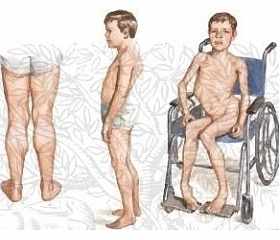
– La debilidad muscular se manifiesta en los miembros inferiores; es proximal, simétrica y progresiva. En etapas posteriores involucra otros grupos musculares, incluyendo los miembros superiores, y hay atrofia.
– El Signo de Gowers es típico: Al pararse desde el piso o la posición sentada, el niño se apoya con las manos sobre sus piernas y muslos.
– La hipertrofia (pseudohipertrofia) de los músculos de las pantorrillas es evidente en etapa temprana. No se observan fasciculaciones, ni pérdida de la sensibilidad.
– Los reflejos osteotendinosos están ausentes o disminuidos.
– Presentan Hiperlordosis Lumbar y abdomen protuberante; luego, Escoliosis Dorso-Lumbar.
– La marcha es inestable, "patoja", con pasos cortos, y aumento de la base de sustentación.
– Cerca de 30% de los pacientes tiene retardo mental (CI menor de 85) no progresivo, retardo del lenguaje y/o dificultad para el aprendizaje, debidos a una variante de la Distrofina, y no sólo por la falta de escolaridad.
– Los hallazgos a la Electromiografía son compatibles con Miopatía, y no específicos.
– La Histopatología (Biopsia Muscular) con tinción normal, revela alteraciones miopáticas inespecíficas, que cambian mientras progresa la enfermedad. La Inmunohistoquímica, más específica, detecta la ausencia de Distrofina en las miofibrillas
– Las enzimas musculares están elevadas, especialmente la Creatin Fosfo Kinasa (CPK) sérica, más de 10 veces su valor normal. Las cifras elevadas son evidentes en etapa preclínica y aún en el Recién Nacido. Descienden con el progreso de la enfermedad.
– Los pacientes sin tratamiento medicamentoso, caminan con apoyo hasta aproximadamente los 12 años. Entonces requieren de sillas de ruedas, y pueden aumentar las contracturas musculares, deformidades de la columna vertebral y la restricción respiratoria.
– La combinación de los antecedentes familiares, edad de comienzo, hallazgos clínicos y su evolución, CPK elevada y los estudios paraclínicos alterados en un varón, son indicativos de DMD.
– Las alteraciones al ECG, arritmias y Miocardiopatía pueden comenzar en la adolescencia, y los pacientes no tratados, generalmente fallecen por Insuficiencia Cardíaca Congestiva y/o Infección Respiratoria alrededor de los 20 años.
Genética
– La DMD es una enfermedad Recesiva Ligada al X.
– El gen normal codifica la Distrofina, ausente en la membrana de las miofibrillas de los pacientes, con formas variantes en cerebro y miocardio.
– El gen denominado 'DMD' es de los más largos encontrados en el genoma humano: tiene unos 2 millones de pares de bases y más de 70 exones.
– Está localizado en Xp21, el brazo corto del cromosoma X, y se han descrito varios genes alelos y más de 5.000 variantes patogénicas.
– La deleción o pérdida parcial de pares de bases es la mutación más frecuentemente encontrada, seguida de la duplicación y las mutaciones de punto. Se detectan por Análisis de ADN, y los resultados deben ser bien interpretados, ya que son el diagnóstico molecular de la DMD. Si no están presentes, no descartan el dx. de DMD; se impone la clínica del paciente.
Asesoramiento genético
– La DMD es una Enfermedad Recesiva Ligada al X. Se presenta en varones: su único cromosoma X tiene el gen mutado. Raras veces en hembras.
– El gen tiene penetrancia completa (100%): Todos los varones con el gen mutado, desarrollan la enfermedad.
– Generalmente, los pacientes fallecen antes de la edad reproductiva, por lo que se considera genéticamente letal.
– Cuando tienen hijos, todos sus varones son sanos (reciben el cromosoma Y del padre); y todas las hijas serán heterocigotas o portadoras (reciben el cromosoma X con el gen mutado del padre).
– Si la madre tiene dos hijos con DMD, o su padre era enfermo, o tiene un sólo hijo enfermo y al menos un hermano (tío materno del propósito) con la enfermedad, es heterocigota o portadora obligada: uno de sus dos cromosomas X tiene la mutación.
– En estos casos no se requiere determinar el gen mutado por Genética Molecular. El Asesoramiento Genético está basado en los antecedentes familiares, en la genealogía.
– Una mujer heterocigota o portadora tiene la probabilidad o riesgo de 25% (1:4) de tener un hijo varón enfermo; 50% (1:2) que su hija sea portadora, como ella; y 50% (1/2), hija homocigota sana.
– Cuando el niño es el único con DMD en la familia (caso único o esporádico), es necesario determinar si la madre es o no portadora, para dar Asesoramiento Genético razonado y preciso.
– Se ha estimado que 2/3 partes de las madres de casos únicos son portadoras de una deleción parcial y en 1/3 de ellas, el hijo enfermo es producto de una mutación nueva o neomutación en el cromosoma X. En éstas, la probabilidad o riesgo de recurrencia es prácticamente 0 (cero).
– La madre del caso único debe ser interrogada y examinada detenidamente, porque algunas portadoras presentan síntomas y signos de la enfermedad, CPK basal elevada, o posterior al ejercicio. Son portadoras sintomáticas.
– En ellas, en las portadoras sanas, asintomáticas, y el hijo enfermo, se requiere el Análisis de ADN (Genética Molecular) para dar Asesoramiento Genético preciso.
– En algunos casos es posible realizar Diagnóstico Prenatal en el 1er. trimestre del embarazo, o Diagnóstico Prenatal no invasivo, y diagnóstico del Recién Nacido.
Tratamiento
– Por la efectividad comprobada en largas series de pacientes, la Prednisona a dosis diarias o inter-diarias es el tratamiento de elección. Aumenta objetivamente la masa y la fuerza muscular, al igual que la función pulmonar, y los pacientes logran caminar independientemente varios años más.
– Se recomienda el uso de L-Carnitina.
– Otros medicamentos, varios tipos de Terapia Génica y de Terapia Celular, no han dado los resultados esperados en estudios clínicos controlados.
– Los pacientes deben ser evaluados regularmente por un Equipo Multidisciplinario bien coordinado.
– Son importantes la alimentación completa, balanceada, suplementos de Vitamina D y Calcio, evitar el sobrepeso y controlar la presión arterial.
– Igualmente, se debe explicar y educar a los padres acerca de la causa de la enfermedad, el cuidado y la evolución de sus hijos enfermos.
– La natación, caminatas, ejercicios, Fisioterapia, Terapia Ocupacional, Terapia de Lenguaje, ejercicios de estiramiento muscular, atención ortopédica y cardiorespiratoria son fundamentales.
– Igualmente, el apoyo del grupo familiar, las actividades educativas, manuales, recreativas y de distracción dentro y fuera del hogar, y la atención Psicopedagógica y Psicológica oportunas, para mejorar su adaptación y calidad de vida.
– Es conveniente que los padres participen y compartan sus experiencias con otros padres de niños enfermos, en grupos de apoyo formales o informales.
Prevención de algunas complicaciones
– Controles y evaluaciones periódicas con los profesionales del Equipo Multidisciplinario.
– Alimentación completa y balanceada, evitando el sobrepeso.
– Suplementos con Vitamina D y Calcio, para evitar Osteopenia y Fracturas.
– Inmunizaciones completas, especialmente las vacunas del Neumococo y de la Influenza Estacionaria.
– Actividades manuales, intelectuales y recreativas, así como el descanso y la relajación, importantes para la salud mental del paciente.
– Ejercicios de estiramiento muscular para evitar las contracturas.
– Caminatas y ejercicios aeróbicos al aire libre, al sol, para mantener el tono y fuerza musculares, mejoran la función pulmonar y cardíaca. Dan bienestar físico, mental y emocional a los pacientes.
(*) El Dr. Frank G. Hammond F. es médico genetista, investigador en la Universidad Centroccidental Lisandro Alvarado.
Imagen de cabecera: levantatuvoz.es.
Fuente: Dr. Frank G. Hammond F. (*) – ucla.edu.ve